QMS Creation and Maintenance Under the IVDR

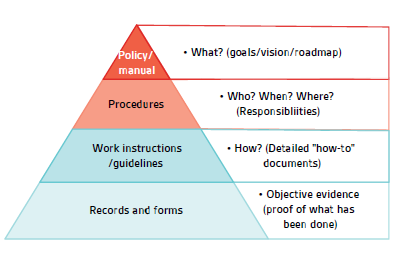
Article 10 (8) of the IVDR establishes a number of requirements and expectations for the QMS of which the most basic is for the manufacturer to establish a QMS. As a reminder, the QMS is made up of all of the documentation that can support the process: A Quality Manual, Quality Policy, Procedures, Work Instructions and Records (see Figure 1). The first part of Article 10 (8) addresses the obvious need to have processes to ensure that production is done in such a way that the device continues to meet the requirements of the regulation throughout the lifetime of the device. This typically includes aspects such as process validation, production process controls and quality controls. For most existing manufacturers, these expectations have neither significantly changed since the previous directive, nor from any other standards. As previously, the QMS should address the process risks and ensure that adequate controls are in place around identified risks. It should also ensure that adequate and accurate records are produced in order to demonstrate and record that the process and products are operating per expectations.
Figure 1 – Documentation pyramid.
While the main requirement of effectively creating, documenting and following a QMS is not substantially different than the previous directive or standards, it is important to note that the IVDR specifically includes the three phrases:
- ‘maintain’,
- ‘keep up to date’ and
- ‘continually improve’.
Since they are generally accepted as synonyms, there must be some significance in the inclusion of both ‘maintain’ and ‘keep up to date’. This brings to mind the notion of ‘timeliness’ and the idea that the QMS must be regularly updated to take into account any sort of changes, whether they are external changes to standards or specifications, internal changes to the organization or products or the results of a process to continuously review and improve the QMS. Later in the phrase we also find the phrase ‘in the most effective manner’, meaning that an overly complicated or complex QMS may be burdensome for the manufacturer to both implement and maintain, as well as possibly be considered non-compliant.
With this in mind, the IVD manufacturer must ensure that an adequate process for reviewing, assessing, documenting and integrating design changes, including changes to the raw materials of the product, is in place. While this may be evident for most manufacturers, this phrase further includes requirements to take into account changes to harmonized standards or common specifications which were used in the development or production of the device. Additionally, there is an emphasis that these must be taken into account in a timely manner. To achieve this, the QMS must then specify not only the harmonized standards and common specifications applicable to a given product, but must also have a process in place to be alerted to changes when they happen (timely), to perform an assessment of those changes and to determine how they impact the product and QMS.
A good monitoring process, which is necessary to identify these kinds of changes, will include various activities such as regulatory surveillance, standards monitoring and review of public findings (warning letters, injunctions). There are many options for implementing this kind of process ranging from companies which offer this kind of assessment as a service, participating in external industry working groups, or setting alerts on websites for key words or subscribing to various newsletters on key topics. People need to have this activity identified as part of their job responsibilities, with adequate time allocated to the process. When changes are identified, a formal assessment should be done to determine what actions will be put into place to account for the new or modified requirements. This documented assessment should include those responsible for performing the assessment (job title, profile or qualifications), as well as those in management who are making or approving the final decision. It is also important to document when a change is determined to not require changes to the QMS to demonstrate that adequate due diligence was done. In the case that changes are extensive, the manufacturer should document the plan for implementing the changes, indicating the expected timelines, along with an explanation of needed tasks. This will provide a means to justify gaps between current standards and those referenced in the QMS if necessary and will ensure alignment within the organization on the expected outcomes.
This is an excerpt from the white paper Developing and maintaining a quality management system for IVDs. To download our other medical device white papers, please visit the Insight page on the Compliance Navigator website.
Request more information today for a call back from a member of our sales team so that you can get a better understanding of how Compliance Navigator can meet your needs.
The Compliance Navigator blog is issued for information only. It does not constitute an official or agreed position of BSI Standards Ltd or of the BSI Notified Body. The views expressed are entirely those of the authors.
