Changes in the Medical Devices Regulation affect drug delivery devices
The Medical Devices Regulation introduces significant changes in approval process under the Medicinal Products Directive
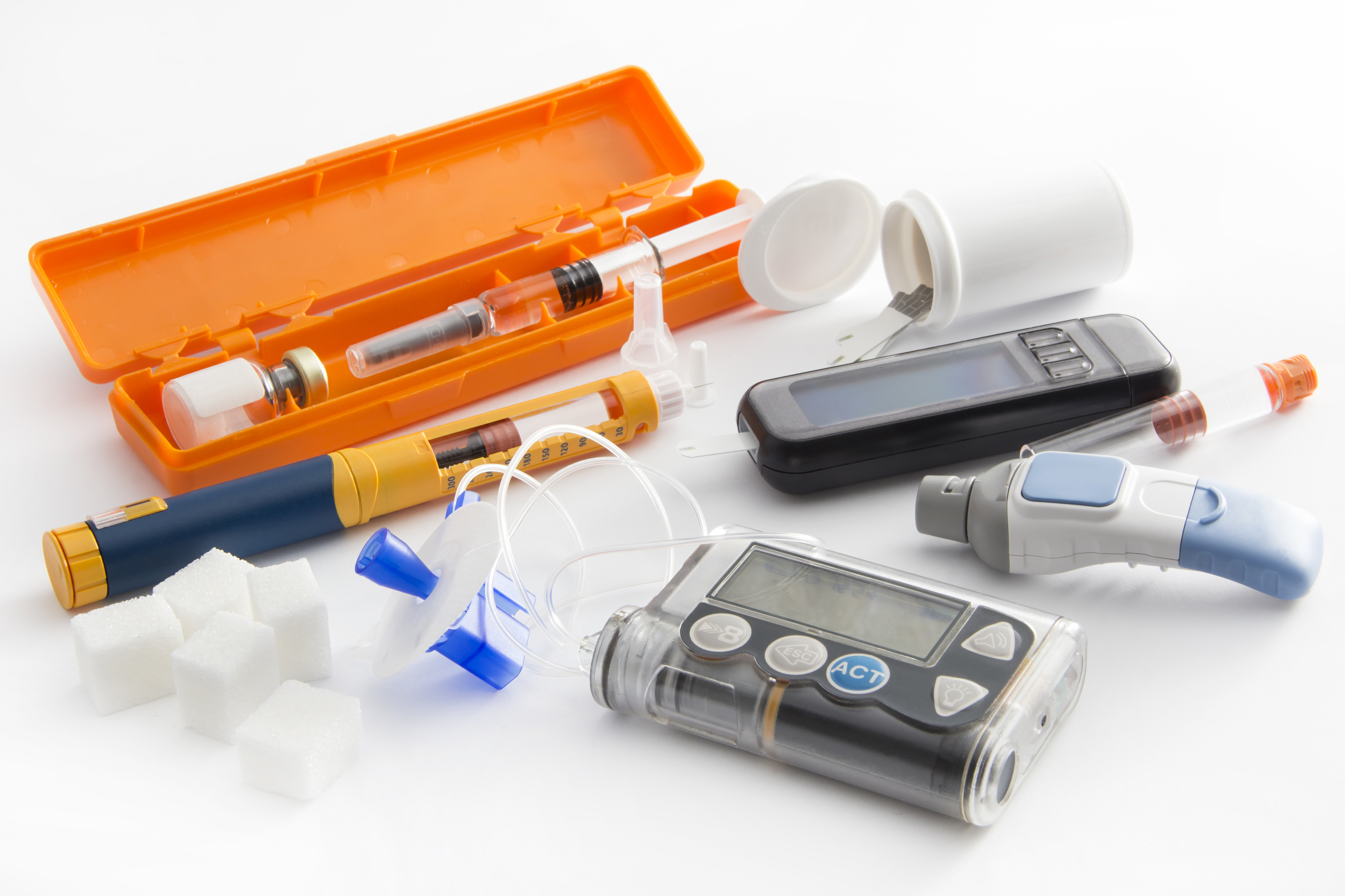
Article 117 of the Medical Devices Regulation (MDR) (Regulation (EU) 2017/745) amends the Directive on medicinal products for human use (MPD) (Directive 2001/83/EC). This will significantly affect pharmaceutical manufacturers supplying drug delivery devices in combination with their medicinal products (such as pre-filled syringes). It will also impact device manufacturers supplying these drug-delivery devices to pharmaceutical manufacturers for inclusion in medicinal products (such as empty syringes). Notified bodies will also be affected.
This change affects medicinal products that incorporate a device that would be covered by the MDR if supplied separately. The dossier for a marketing authorisation under the MPD will have to include evidence of the conformity of the device part with the applicable general safety and performance requirements in Annex I to the MDR. This could be either:
- an EU declaration of conformity or the relevant certificate issued by a notified body that allows a CE mark to be affixed to the device, or
- an opinion on the conformity of the device with the general safety and performance requirements in the MDR. This has to be issued by an appropriately-designated notified body, if the conformity assessment of the device, if used separately, requires the involvement of a notified body.
Manufacturers who are developing novel drug delivery devices need to consider this change in requirements and assess the impact on their development programmes. If they do not intend to place their devices on the market separately from the medicinal product, they need to either obtain certification that would enable CE-marking of the device or identify a notified body to provide an opinion on the conformity of the device to the MDR.
Device manufacturers supplying drug-delivery systems to the pharmaceutical industry are likely to be asked to provide copies of their declaration of conformity or obtain opinions from a notified body on the conformity of the device to the MDR.
Notified bodies should be aware that they will be approached to provide opinions on conformity of drug-delivery devices that are not CE-marked. They should consider if they are able to provide this service and what type of documentation they will use to provide their opinion.
Author: Eamonn Hoxey, of E V Hoxey Ltd, UK, is a writer, trainer and consultant on a range of life science areas including regulatory compliance, quality management, sterility assurance and standards development.
The Compliance Navigator blog is issued for information only. It does not constitute an official or agreed position of BSI Standards Ltd or of the BSI Notified Body. The views expressed are entirely those of the authors.
